
Неделя оглашения лауреатов Нобелевской премии 2024 года оказалась выдающейся. Премия по физике присуждена за разработку алгоритмов искусственного интеллекта на основе нейронных сетей, что подчеркнуло их огромное значение для науки и общества. На следующий день мир узнал, что Нобелевскую премию по химии за 2024 год получат Дэвид Бейкер (David Baker), Демис Хассабис (Demis Hassabis) и Джон Джампер (John Jumper) «за вычислительный дизайн белков и предсказание их структуры». Две премии связаны: если «физическая», по сути, присуждена за создание принципиального нового инструмента, то «химическая» — за полученный с помощью этого инструмента выдающийся результат. Разработанная Бейкером с коллегами программа Rosetta позволила ученым создать принципиально новые (дизайнерские) белки с нужной трехмерной структурой. Между тем алгоритмы Джампера и Хассабиса несколько лет назад достигли небывалой точности предсказания структуры белков на основе аминокислотной последовательности. Теперь их детище AlphaFold2 не уступает дорогостоящим «мокрым» (то есть экспериментальным) методам анализа белка вроде рентгеноструктурного анализа и ядерного магнитного резонанса.
Белки в сиянии Нобелевской медали
Белковые молекулы играют ключевую роль в живом организме — ученые знали об этом довольно давно. В качестве самостоятельного класса биологических молекул их выделил Антуан де Фуркруа еще на заре современной химии, в 1789 году. Столетие спустя Фридрих Энгельс определил жизнь как «способ существования белковых тел». Однако понимание того, что же такое есть белки, как они работают и как могут быть использованы, потребовало многих десятилетий работы. Теперь, после ряда эпохальных открытий мы можем сказать, что достаточно хорошо разобрались со структурой и функциями белков. Это подчеркивает Нобелевская премия по химии этого года, лауреаты которой уже известны, а вручение состоится 10 декабря в Стокгольме. Половина награды присуждена Дэвиду Бейкеру (David Baker) из Вашингтонского университета и Медицинского института Говарда Хьюза (США), еще по четверти достанется за общие работы Демису Хассабису (Demis Hassabis) и Джону Джамперу (John M. Jumper) из Google DeepMind (компании, занимающейся искусственным интеллектом).
Что же такое белки, с которыми достигнут такой впечатляющий прогресс? За последние сто лет удалось выяснить, что эти крупные биологические молекулы представляют собой полимеры — цепочки из повторяющихся блоков-мономеров. В роли мономеров выступают остатки 20 различных аминокислот (в отдельных случаях к ним добавляются селеноцистеин и пирролизин), которые сильно отличаются по размеру, форме, заряду, гидрофобности и другим характеристикам. Их уникальный порядок в полипептидной цепочке определяется информацией, закодированной в ДНК и переданной к рибосоме матричной РНК согласно Центральной догме молекулярной биологии Френсиса Крика. Она была сформулирована благодаря открытию структуры ДНК, за которую Крик, Джеймс Уотсон и Морис Уилкинс получили Нобелевскую премию по физиологии и медицине в 1962 году.
Созданная на рибосоме цепочка самопроизвольно приобретает сложную трехмерную структуру, которая определяется только порядком аминокислот в ней. Этот принцип сформулировал Кристиан Анфинсен (его иногда называют «догмой Анфинсена»), за что был удостоен Нобелевской премии по химии в 1972 году. Если белок денатурирует, то есть теряет свою нормальную форму (например, когда мы варим или взбиваем яичный белок), он перестает выполнять свои функции. Анфинсен показал, что в ряде случаев денатурация обратима — противоположный процесс ренатурации возвращает белку нужную структуру и функции. Например, фермент может снова делать свою работу — избирательно ускорять нужные химические реакции. Опять-таки, ренатурация происходит на основе аминокислотной последовательности белка, которую называют «первичной структурой».
Строение белковой молекулы сложно и своеобразно: она отличается от «обычных» высокомолекулярных соединений и живет по своим законам. Для белка характерна иерархия уровней структурной организации (рис. 1). Первый уровень — это и есть цепочка соединенных в определенном порядке, но обычно без правильного чередования аминокислотных остатков — полипептидная последовательность. Впервые первичную структуру белка (а именно — инсулина) установил Фредерик Сенгер, за что был отмечен Нобелевской премией по химии в 1958 году.
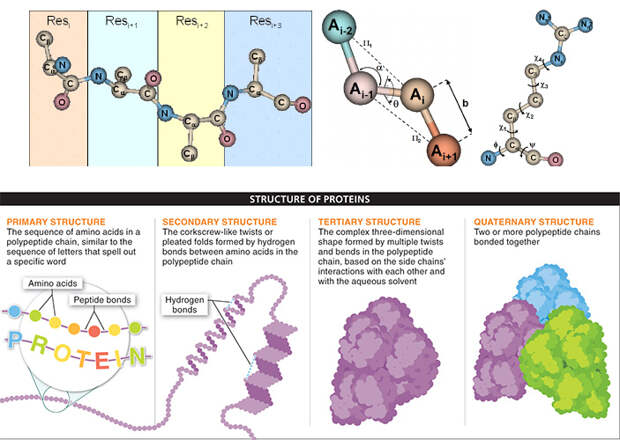
Строение белковой молекулы сложно и своеобразно: она отличается от «обычных» высокомолекулярных соединений и живет по своим законам. Для белка характерна иерархия уровней структурной организации (рис. 1). Первый уровень — это и есть цепочка соединенных в определенном порядке, но обычно без правильного чередования аминокислотных остатков — полипептидная последовательность. Впервые первичную структуру белка (а именно — инсулина) установил Фредерик Сенгер, за что был отмечен Нобелевской премией по химии в 1958 году.
Расположенные рядом аминокислоты самопроизвольно образуют устойчивые блоки вторичной структуры, которые удерживаются водородными связями. Это — альфа-спирали и бета-слои. Помимо них белок может также иметь более подвижные петли. Структуру альфа-спирали первым расшифровал (или, как говорят химики, решил) Лайнус Полинг — он стал лауреатом Нобелевской премии по химии 1954 года.
Блоки вторичной структуры сложным образом взаимодействуют между собой за счет различных механизмов — ковалентных (дисульфидные мостики), ионных и водородных связей, а также гидрофобных взаимодействий. В результате молекула приобретает характерную общую форму — третичную структуру, которая обычно строго фиксирована. Настолько строго, что многие белки успешно превращают в кристаллы с одинаковым взаиморасположением молекул относительно друг друга. Именно их изучение (кристаллография) помогло Максу Перуцу и Джону Кендрю, получившим Нобелевскую премию по химии в 1962 году, установить первую полную структуру белка — гемоглобина. Два года спустя высшей научной премией была награждена Дороти Ходжкин, которая использовала тот же метод для изучения важных для биохимии молекул, в том числе инсулина. Наконец, некоторые белки работают не как отдельные молекулы из одной цепочки аминокислот, а в виде комплекса молекул. В состав комплекса могут входить два, три или более белков и другие молекулы, — такие белки имеют четвертичную структуру (рис. 1).
Процесс приобретения белком уникальный формы называется «фолдинг» (то есть сворачивание). То, как это происходит, — самостоятельный вопрос биофизики, сложность которого сформулировал в форме парадокса Сайрус Левинталь: для белка размером всего 100 аминокислотных остатков на поиск нужной трехмерной формы путем полного их перебора потребуется 1080 лет. Это многократно превышает время существования Вселенной, тогда как белки сворачиваются и ренатурируют за ничтожные доли секунды. Сейчас мы знаем, что на пути к нужной структуре белок постепенно опускается в воронку на «энергетическом ландшафте», где находятся близкие к нему формы, и постепенно организуется из полурасплавленного состояния (molten globule; подробнее см. Фолдинг белков: 2. Главная и боковые цепи)
Результатом успешного «поиска себя» белковой молекулы становится специфичная структура, которая неразрывно связана с функциями белка. В первом приближении белки делят на фибриллярные (длинные волокна, нерастворимые в воде, которые часто образуют опорные структуры), глобулярные (имеют округлую форму и хорошо растворяются в воде) и мембранные (работают в мембранах, в которые погружена их гидрофобная часть). Белковые молекулы выполняют большинство разнообразных биологических функций, в том числе катализируют химические реакции (такие белки называют ферментами), обеспечивают экспрессию генов, придают клетке нужную структуру и делают ее и живой организм в целом управляемыми. Нарушения работы отдельных белков лежат в основе многих болезней, а придание молекулам нужных функций особенно важно для биотехнологий, — однако раньше решение таких вопросов упиралось в неизвестную структуру белка.
Расшифровка «языка белков»
Итак, трудами Перутца, Анфинсена и многих других ученых было показано, что аминокислотная последовательность белка может быть определена и напрямую связана с его трехмерной структурой, которую тоже научились устанавливать экспериментально (хотя и ценой больший усилий: порой — десятилетий работы). Оставалось понять, как последовательность и структура белка связаны между собой, и создать модели с достаточной предсказательной силой. Было понятно, что задача имеет исключительное значение и потому привлекла внимание многих ученых. Венкатраман Рамакришнан, — лауреат Нобелевской премии по химии 2009 года за исследования рибосом, обеспечивающих биосинтез белка (совместно с Томасом Стейцем и Адой Йонат) — назвал ее «великим вызовом биологии с полувековой историей». Решить настолько фундаментальную проблему удалось далеко не сразу.
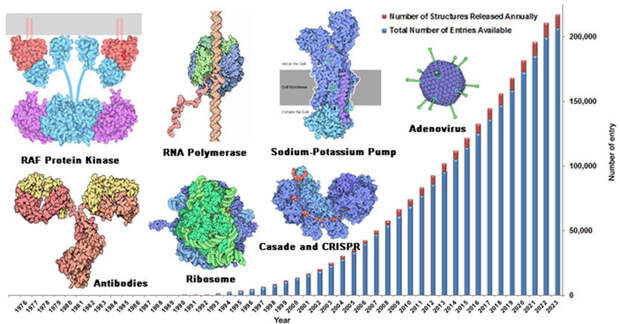
Постепенно развивались новые методы биологических исследований, которые могут способствовать прогрессу науки не меньше, чем самые великие теории. К 1990-м годам большие основным методом исследований стал рентгеноструктурный анализ (РСА), использующий кристаллы белковых молекул, чтобы по картине дифракции рентгеновских лучей понять их строение. В результате быстро копились решенные структуры белков, разрешение метода росло. Затем к РСА добавились другие способы их получения, — спектроскопия ядерного магнитного резонанса (ЯМР) и криоэлектронная микроскопия (см. Нобелевская премия по химии — 2017, «Элементы», 12.10.2017). Структуры белков собирали в базы данных — прежде всего самую крупную PDB (Protein Data Bank), которая существует с 1970-х годов. Долгое время PDB существовал исключительно оффлайн (в частности, в виде записей на магнитных лентах), однако с появлением интернета стал доступен широкому кругу ученых. С началом XXI века объемы PDB стали очень быстро расти и сейчас число расшифрованных структур молекул белков приближается к 200 тысячам (рис. 2).
С другой стороны, в конце XX века также развивались методы секвенирования ДНК, то есть установления ее первичной структуры (последовательности нуклеотидов). Появилось секвенирование нового поколения (next generation sequencing, NGS) на основе различных платформ. В результате стоимость «прочтения» нуклеиновых кислот многократно снизилась, а число известных последовательностей лавинообразно нарастало. Среди них многие представляют собой кодирующие последовательности генов — именно они (при участии РНК) служат матрицей для синтеза белков на рибосоме. Так с помощью простого перевода на «белковый язык» стало возможным получение данных о последовательности огромного числа полипептидов.
Тем временем подоспели и алгоритмы искусственного интеллекта (artificial intelligence, AI), которые уже используются во многих областях деятельности, в том числе — в науке. Это стало возможным благодаря быстрому прогрессу вычислительной техники. Здесь стоит выделить искусственные нейронные сети, разработкой которых начиная с 1980-х годов занимались нобелевские лауреаты 2024 года по физике — изобретатель модели ассоциативной памяти Джон Хопфилд (John Hopfield) и создатель алгоритмов «глубокого обучения» Джеффри Хинтон (Geoffrey Hinton). Не все согласны, что их работы относятся к области физики, но значимость нейросетей очевидна любому. Символично, что имена Бейкера, Хассабиса и Джампера, которые сумели найти искусственному интеллекту одно из самых плодотворных и важных применений, были названы нобелевским комитетом ровно день спустя после имен «физиков».
Удивительно, что среди двух тесно связанных задач — предсказание структуры существующих белков и создание новых (дизайнерских) белков — раньше была решена вторая. По-видимому, это связано с исходно заданной третичной структурой при белковом дизайне, на которую проще опираться. Также в этом случае нет ограничений по последовательности — ученые и их программы вольны «творить» дизайнерский белок из любых аминокислотных звеньев. Первые создаваемые de novo (то есть «с нуля») белки получали без помощи вычислительной техники. Ученые руководствовались знаниями о принципах организации белка, закономерностях структур молекул и особенностях аминокислот в их составе. Первыми стали Линн Реган (Lynne Regan) и Уильям ДеГрадо (William F. DeGrado), создавшие в 1988 году новый простой белок из четырех альфа-спиралей (L. Regan, W. DeGrado, 1988. Characterization of a helical protein designed from first principles), а также Олег Борисович Птицын и его коллеги, которые в 1992 году синтезировали белок с альфа-спиралями и бета-слоями — альбебетин (A. Fedorov et al., 1992. De novo design, synthesis and study of albebetin, a polypeptide with a predetermined three-dimensional structure: Probing the structure at the nanogram level). Компьютерное моделирование к задаче впервые применили в 1997 году, превратив белок с мотивом «цинковый палец» в очень похожую по форме молекулу, не требующую для стабилизации ионов цинка (B. Dahiyat, S. Mayo, 1997. De Novo Protein Design: Fully Automated Sequence Selection). При этом последовательности нового и старого белка были идентичны лишь на 21%.
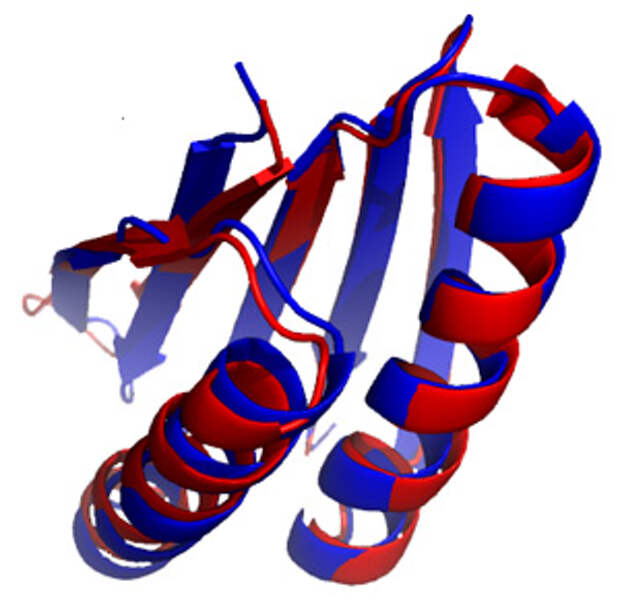
Настоящий прорыв в белковом дизайне случился в 2003 году и связан с именем Дэвида Бейкера и его коллег. Им удалось создать белок Top7, который состоял из 93 аминокислотных остатков, образующих два бета-слоя и одну альфа-спираль. Соответствие структуры синтезированной молекулы с исходным замыслом подтвердили с помощью РСА — на месте кроме остова оказались все аминокислотные остатки довольно крупной молекулы (рис. 3). При этом образованный блоками вторичной структуры паттерн (белковая укладка) и последовательность Top7 были уникальными — похожих нет ни у одного «природного» белка. Это в полном смысле слова новый и уникальный белок, который, однако, не способен выполнять какой-либо полезной функции.
Успех стал возможен благодаря программе Rosetta, созданной Бейкером и его командой в 1999 году и названной в честь розеттского камня — того, что помог расшифровать первые египетские иероглифы. Rosetta объединяла вместе короткие последовательности от разных белков из PDB и оптимизировала их одновременно с предполагаемой структурой так, чтобы та соответствовала заданной модели. Такая блочная сборка — главная идея в основе Rosetta. Для оценки энергии структуры использовались методы Монте-Карло и учитывались силы Ван-дер-Ваальса, водородные связи и роль растворителя. Rosetta последовательно получала множество возможных структур, ранжированных по своей энергии, то есть «качеству». Так Бейкер и его коллеги создали множество новых белковых структур. Помимо дизайна белков Rosetta стали применять для дизайна их новых функций. В 2008 году коллектив сообщил о первых попытках de novo синтеза фермента, способного катализировать реакции, которые не ускоряют известные белки (L. Jiang et al., 2008. De Novo Computational Design of Retro-Aldol Enzymes). Молекулы был синтезированы, однако сильно уступали по эффективности природным ферментам. Вероятно, улучшить работу дизайнерских ферментов в будущем удастся с помощью направленной эволюции (по-английски directed evolution). Гораздо успешнее прошел синтез новых лиганд-связывающих белков, с высокой специфичностью узнающих стероиды (вплоть до пикомолярной концентрации, порядка 10−12 моль; C. Tinberg et al., 2013. Computational design of ligand-binding proteins with high affinity and selectivity).
Стоит отметить, что Rosetta годится не только для белкового дизайна, но и для решения обратной задачи, — предсказания третичной структуры по аминокислотной последовательности. Несмотря на определенный успех Rosetta, настоящую революцию в этой области произвели другие люди и другие программы.
Предсказания сбываются
На рубеже веков, когда были готовы предпосылки грядущей «белковой революции» — PDB, NGS и AI, — предсказание структуры белков простимулировал один очень значимый конкурс. CASP (Critical Assessment of protein Structure Prediction — «критическая оценка предсказания белковых структур») проходит раз в два года начиная с 1994 года и сейчас собирает более сотни коллективов ученых. Они используют различные алгоритмы, чтобы как можно точнее предсказать трехмерную структуру новых белков и превзойти в этом конкурентов. Соревнование отличается особой объективностью: на момент создания участниками моделей никто (включая организаторов) не знает, как в действительности выглядят целевые белки. Обычно их структура еще даже не решена — по крайней мере, не отправлена в PDB. Точность предсказаний оценивают как расстояния между парами соответствующих Сα-атомов (принятые обозначения атомов в полипептидной цепи, см. рис. 1) в структурном выравнивании модели и реального белка.
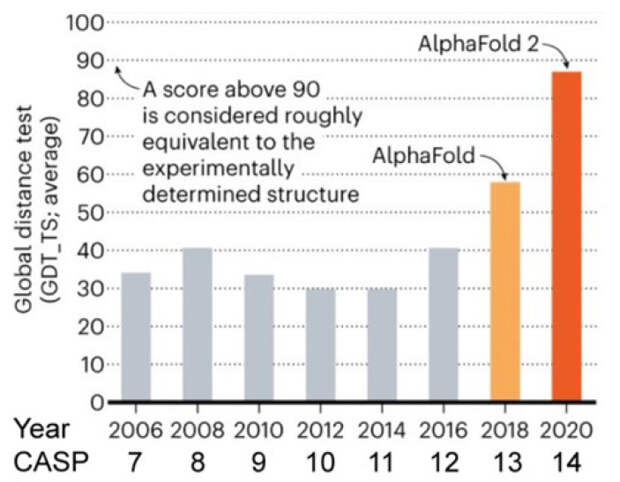
Долгое время участники CASP не могли похвастаться высокой точностью предсказаний — она достигала как максимум 40%. Перелом наступил в 2018 году на CASP13, когда компания DeepMind во главе с Демисом Хассабисом использовала программу, основанную на сверточных нейронных сетях (convolutional neural network) — AlphaFold, ныне известную как AlphaFold1 или AF1. Она создавала двухмерную матрицу расстояний для отдельных аминокислотных остатков и использовала иерархическое извлечение паттернов, который ранее применяли к задачам распознавания образов (image recognition). Именно их команда тогда победила — с рекордными значением точности, почти 60%. Любопытно, что Хассабис и его команда к тому времени уже добились впечатляющих успехов в играх с участием ИИ. В 2016 году их программа AlphaGO одержала верх над одним из самых сильных игроков в го — Ли Седолем, что заставило его закончить карьеру. Другие детища DeepMind, также основанные на глубоком обучении (deep learning), были очень успешны в шахматах и компьютерных играх.
Следующее соревнование в 2020 году не только закрепило успех команды DeepMInd, но и показало новый уровень их алгоритмов. Программа AlphaFold2 была не просто усовершенствована, но качественно переработана. Ее основой стали не сверточные сети, а архитектура Transformer, в результате чего точность предсказаний достигала 90%. То есть она была сравнима с ранее не имевшими альтернативы экспериментальными методами, гораздо более трудоемкими и дорогими. Это сделало возможным точное предсказание структуры огромного количества белков, — по сути всех, гены которых секвенированы и для которых известна аминокислотная последовательность. Вскоре AlphaFold2 использовали для получения трехмерных моделей всех белков человека, затем — для решения свыше 200 миллионов различных белковых молекул.

Работа AlphaFold2 основана на двух блоках — эвоформере (evoformer) и структурном модуле (structure module; см. рис. 5). Первый блок оперирует одновременно двумя представлениями белковых последовательностей: двумерной матрицей расстояний для множественного выравнивания белков разных видов живых существ, а также матрицей попарных расстояний. Два представления данных постепенно меняют друг друга в ходе процесса обучения алгоритмов и «эволюционируют». Далее структурный блок работает напрямую с трехмерным остовом («скелетом») получающегося белка, сравнивая его с целевой структурой. Остов при этом моделируют набором треугольников, образованный атомами N, Cα и C каждого из аминокислотных остатков (рис. 5, обозначения атомов см. на рис. 1). Треугольники находятся в свободном движении в виде своеобразного «аминокислотного газа». Система итеративно (то есть раз за разом) обновляется за счет разворотов и перемещений треугольников с помощью механизма инвариантной точки внимания (invariant point attention). Наконец, происходит предсказание углов, которые описывают вращение вокруг связи между соседними аминокислотными остатками, что позволяет добавить к модели их координаты. Получившаяся структура возвращается назад (в блок-эвоформер) для новых циклов оптимизации — они повторяются до тех пор, пока предсказание не приобретет нужные показатели качества.
Незадолго до триумфа AlfaFold2 и его создателей, в мае 2024 года стало известно о создании AlphaFold3. Это новый шаг вперед в белковых предсказаниях. Известно, что программа AlphaFold3 преодолела ограничение предшественниц и способна предсказывать взаимодействия между белковыми молекулами, а также структуры их комплексов с ДНК, РНК, ионами и другими лигандами.

По нобелевским меркам премия нашла своих лауреатов довольно скоро — с момента победы AlphaFold2 на конкурсе прошло всего несколько лет. Создателю Rosetta, впрочем, пришлось ждать заметно дольше. Тем не менее высшая награда научного мира указывает на огромное значение алгоритмов, с которыми работали Бейкер, Хассабис и Джампер. Совсем недавно и «по-отдельности» они были также удостоены престижного научного приза Breakthrough Prize. Их работа уже принесла первые впечатляющие и несомненно практические результаты. С помощью Rosetta созданы новые наноматериалы, молекулярные роторы, вакцины и ингибиторы вирусных частиц, сенсоры, которые способны замечать в окружающей среде опасные вещества, и другие полезные молекулы (рис. 6). Огромный поток точно предсказанных с помощью AlphaFold2 структур еще ждет своего часа. Наверняка они помогут понять молекулярные основы многих болезней, в том числе редких генетических (орфанных), и простимулируют развитие биомедицины и технологии. Значимость этого научного очевидно велика, однако оценить ее в полной мере станет возможно только спустя годы.
Михаил Орлов
Свежие комментарии